
Polymerase chain reaction (PCR) is a relatively simple and widely used molecular biology technique to amplify and detect DNA and RNA sequences. Compared to traditional methods of DNA cloning and amplification, which can often take days, PCR requires only a few hours. PCR is highly sensitive and requires minimal template for detection and amplification of specific sequences. Basic PCR methods have further advanced from simple DNA and RNA detection. Below, we have provided an overview of the different PCR methods and the reagents we provide at Enzo Life Sciences for your research needs. We aim to help scientists quickly access PCR reagents to use in their next research project!
PCR
For standard PCR, all you need is a DNA polymerase, magnesium, nucleotides, primers, the DNA template to be amplified and a thermocycler. The PCR mechanism is as simple as its purpose: 1) double-stranded DNA (dsDNA) is heat denatured, 2) primers align to the single DNA strands and 3) the primers are extended by DNA polymerase, resulting in two copies of the original DNA strand. The denaturation, annealing, and elongation process over a series of temperatures and times is known as one cycle of amplification. Each step of the cycle should be optimized for the template and primer set used. This cycle is repeated approximately 20-40 times and the amplified product can then be analyzed. PCR is widely used to amplify DNA for subsequent experimental use. PCR also has applications in genetic testing or for the detection of pathogenic DNA.
As PCR is a highly sensitive method and very small volumes are required for single reactions, preparation of a master mix for several reactions is recommended. The master mix must be well mixed and then split by the number of reactions, ensuring that each reaction will contain the same amount of enzyme, dNTPs and primers. Many suppliers, such as Enzo Life Sciences, also offer PCR mixes that already contain everything except primers and the DNA template.
Guanine/Cytosine-rich (GC-rich) regions represent a challenge in standard PCR techniques. GC-rich sequences are more stable than sequences with lower GC content. Furthermore, GC-rich sequences tend to form secondary structures, such as hairpin loops. As a result, GC-rich double strands are difficult to completely separate during the denaturation phase. Consequently, DNA polymerase cannot synthesize the new strand without hindrance. A higher denaturation temperature can improve this and adjustments towards a higher annealing temperature and shorter annealing time can prevent unspecific binding of GC-rich primers. Additional reagents can improve the amplification of GC-rich sequences. DMSO, glycerol and betaine help to disrupt the secondary structures that are caused by GC interactions and thereby facilitate separation of the double strands.
Hot Start PCR
Unspecific amplification is a problem that can occur during PCR. Most DNA polymerases that are used for PCR, work best at 68 – 72°C. Therefore, the chosen extension temperature should be in this range. The enzyme can, however, also be active to a lesser degree, at lower temperatures. At temperatures that are far below the annealing temperature, primers tend to bind non-specifically, which can lead to non-specific amplification, even if the reaction is set up on ice. This can be prevented by using polymerase inhibitors that dissociate from the DNA polymerase only once a certain temperature is reached. The inhibitor can be an antibody that binds the polymerase and denatures at the initial denaturation temperature.
RT-PCR
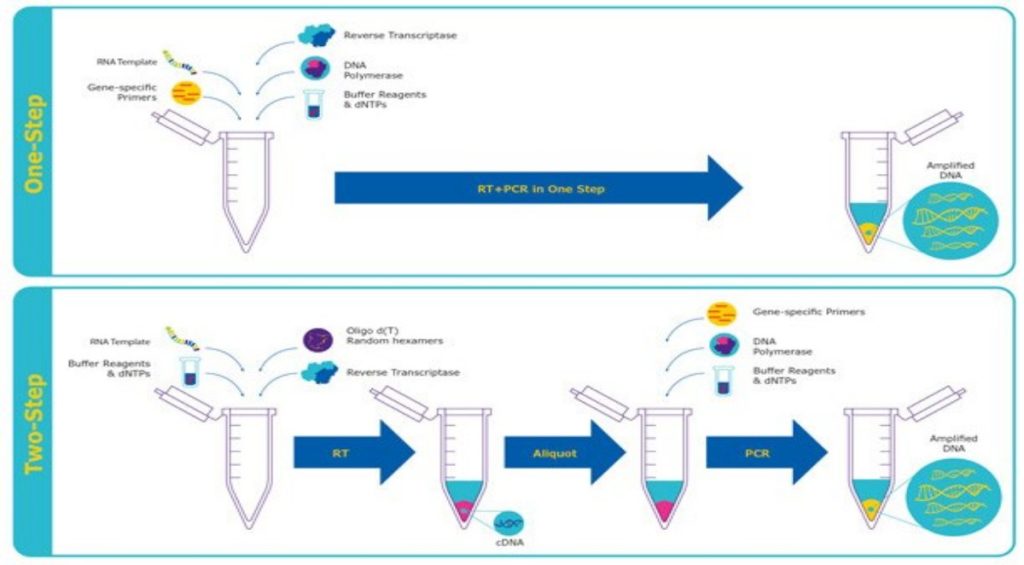
Reverse transcription PCR, or RT-PCR, allows the use of RNA as a template. An additional step allows the detection and amplification of RNA. The RNA is reverse transcribed into complementary DNA (cDNA), using reverse transcriptase. The quality and purity of the RNA template is essential for the success of RT-PCR. The first step of RT-PCR is the synthesis of a DNA/RNA hybrid. Reverse transcriptase also has an RNase H function, which degrades the RNA portion of the hybrid. The single stranded DNA molecule is then completed by the DNA-dependent DNA polymerase activity of the reverse transcriptase into cDNA. The efficiency of the first-strand reaction can affect the amplification process. From here on, the standard PCR procedure is used to amplify the cDNA. The possibility to revert RNA into cDNA by RT-PCR has many advantages. RNA is single-stranded and very unstable, which makes it difficult to work with. Most commonly, it serves as a first step in qPCR, which quantifies RNA transcripts in a biological sample.
qPCR and RT-qPCR
Quantitative PCR (qPCR) is used to detect, characterize and quantify nucleic acids for numerous applications. Commonly, in RT-qPCR, RNA transcripts are quantified by reverse transcribing them into cDNA first, as described above and then qPCR is subsequently carried out. As in standard PCR, DNA is amplified by 3 repeating steps: denaturation, annealing and elongation. However, in qPCR, fluorescent labeling enables the collection of data as PCR progresses. This technique has many benefits due to a range of methods and chemistries available.
In dye-based qPCR (typically green), fluorescent labeling allows the quantification of the amplified DNA molecules by employing the use of a dsDNA binding dye. During each cycle, the fluorescence is measured. The fluorescence signal increases proportionally to the amount of replicated DNA and hence the DNA is quantified in “real time”. The disadvantages to dye-based qPCR are that only one target can be examined at a time and that the dye will bind to any ds-DNA present in the sample.
In probe-based qPCR, many targets can be detected simultaneously in each sample but this requires optimization and design of a target specific probe(s), used in addition to primers. There are several types of probe designs available, but the most common type is a hydrolysis probe, which incorporates the use of a fluorophore and quencher. Fluorescence resonance energy transfer (FRET) prevents the emission of the fluorophore via the quencher while the probe is intact. However, during the PCR reaction, the probe is hydrolyzed during primer extension and amplification of the specific sequence it is bound to. The cleavage of the probe separates the fluorophore from the quencher and results in an amplification-dependent increase in fluorescence. Thus, the fluorescence signal from a probe-based qPCR reaction is proportional to the amount of the probe target sequence present in the sample. Because probe-based qPCR is more specific than dye-based qPCR, it is often the technology used in qPCR diagnostic assays.