Meaning of Disinfectant and Antiseptic:
Disinfectants are antimicrobial agents that are applied to non-living objects to destroy microorganisms. The process of killing the microbes is called disinfection. It may be defined as “cleaning of an article of some or all of the pathogenic organisms that cause infection”.
Antiseptics (from Greek anti- against + septikos – putrefactive) are antimicrobial substances that are applied to living tissue such as skin to reduce the possibility of infection, sepsis, or putrefaction. Some antiseptics are true germicides, capable of destroying microbes (bactericidal), whilst others are bacteriostatic and only prevent or inhibit their growth.
Antibacterial are antiseptics that only act against bacteria. Micro-biocides kill virus particles are called viricides. For practical purposes, antiseptics are routinely categorized as topical agents, for application to skin, mucous membranes, and inanimate (non-living objects).
Although a formal definition includes agents which are used internally such as the urinary tract antiseptics. Bacterial endospores are most resistant to disinfectants; however, some viruses and bacteria also possess some tolerance.
Properties of Disinfectant and Antiseptic:
A perfect disinfectant would offer complete and full sterilization, without harming other forms of life. It should not be expensive, and non-corrosive. Most disinfectants are also, by nature are potentially harmful (even toxic) to humans or animals. They should be used with appropriate care. Most come with safety instructions printed on the packaging.
Most modem household disinfectants contain Bitrex, an exceptionally bitter substance designed to discourage ingestion, as an added safety measure. Those that are used indoors should never be mixed with other cleaning products as chemical reactions may occur. Disinfectants are frequently used in hospitals, dental surgeries, kitchens and bathrooms to kill infectious organisms.
The choice of the disinfectant to be used depends on the particular situation. Some disinfectants have a wide spectrum (kill nearly all microorganisms), whereas others kill a smaller range of disease- causing organisms but are preferred for other properties such as non-corrosive, non-toxic, or inexpensive of nature.
Selection Criteria of Disinfectant and Antiseptic:
Usually disinfectants are ‘cidal’ in that they kill the susceptible potential pathogenic agents. The selection of a disinfectant should be based on the function. The disinfectant should be broad spectrum (eliminates bacteria, viruses, protozoa, fungi and spores), non-irritating, non-toxic, non-corrosive and inexpensive. Selection should be based on effectiveness against the potential pathogenic agent, safety to people, impact on equipment, environment and expenses.
Effectiveness of disinfectant depends on type of contaminating microorganism, degree of contamination i.e. determines the quality of disinfectant required and time of exposure and the amount of proteinaceous material present because high protein based materials absorb and neutralize some chemical disinfectants.
Further, presence of organic matter and other compounds such as soaps may neutralize some disinfectants. Chemical nature of disinfectant is important to understand the mode of action in order to select the appropriate disinfectant.
Besides, concentration and quantity of disinfectant is important to choose the proper concentration and quantity of disinfectant suited to each situation. The time and appropriate temperature must be allowed for action of the disinfectant and may depend on the degree of contamination and organic matter load.
The residual activity and effects on fabric and metal should be considered for specific situations. Application temperature, pH and interactions with other compounds must also be considered. Finally, the consideration may be given on toxicity to the environment and relative safety to people that may be exposed.
Disinfection Kinetics:
When a single unit of microorganisms is exposed to a single unit of disinfectant, the reduction in microorganisms follows a first-order reaction. This relationship between kill, efficiency and contact time, was developed by Harriet Chick.
dN/dt = -kN
N = N0e-kt
This equation is known as Chick’s Law:
N = number of microorganism (N0 is initial number)
k = disinfection constant
t = contact time
Evaluation of Disinfectant:
Evaluation of disinfectant by Phenol coefficient method is described in detail in Practical Microbiology by Dubey and Maheshwari (2007).
Sterility Test:
Sterility testing of pharmaceutical products is carried out during the sterilization-validation process as well as for routine release testing as shown in Fig. 19.6. It determines suitability of a finished product of pharmacopoeial requirements. The understanding of sterility testing is beneficial in terms of designing a validation process. It provides adequate and reliable sterility test data required for quality assurance.
1. Sterility Test Conditions:
The following sterility test conditions are required:
(i) Environmental Conditions:
It should be designated to provide environmental conditions which avoid accidental contamination of the product during the test, i.e. conditions equivalent to those for the aseptic preparation of pharmaceutical products. A suitable environment is a class A laminar airflow cabinet located in a class B room. Regular microbiological monitoring of the working area with contact swabs and settle plates should be carried out.
(ii) Culture Conditions:
Sterility testing involves testing of viable microorganisms on culture media. It follows appropriate conditions for growth microorganism of on culture media. Factors affecting the growth of microorganisms indicate the importance of selecting the most appropriate culture conditions for sterility testing.
(iii) Factors affecting growth of bacteria and fungi:
A number of factors are responsible for growth of microorganisms. The major factors affecting growth are nutrition, moisture, air, temperature, pH, light, osmotic pressure and the presence of growth inhibitors which affect the growth of bacteria and fungi.
(a) Nutrition:
Nutritional substances such as source of carbon, nitrogen, mineral salts and growth factors must be present in the suitable and adequate amount in culture medium to produce vigorous growth of microorganisms. Sometimes, supplementary source of carbohydrate must be added to media because of high concentration requirements.
Some pathogenic fungi grow better in media containing extracts from animal sources such as beef extract. The soya bean casein digest medium used in fungal sterility test contains tryptone (a pancreatic digest of casern) as well as the vegetative extract (soya peptone).
(b) Moisture:
Generally, a medium for the growth of bacteria must contain at least 20% of water. In the absence of moisture, bacteria cease to multiply but spore bearing forms may continue to exist in form of spore for many years. The moisture requirement of fungi is more than bacteria.
(c) Air:
Air affects growth of microorganisms. Many bacteria (e.g. Pseudomonas aeruginosa, E. coli, Serratia mareescaus) and fungi (saprophytic mould such as Mucor, Rhizopus etc.) are aerobic ones. Some microorganisms such as Clostridium tetani are anaerobes that multiply in the absence of air. There is a third group called facultative anaerobes (e.g. yeast) which are able to grow with or without air.
(d) Temperature:
Most pathogenic bacteria multiply at normal human body temperature, i.e. at 37°C. Few are serious and common contaminant of wounds, eye drops and injections (Pseudomonas spp.).
They have an optimum growth temperature of 30°C and may not be detected at 37°C. Optimum temperatures for growth of most moulds and yeast lie between 20 and 25°C. As many moulds grow rather slowly plates should be incubated for at least 14 days. Few bacteria are thermophiles that optimum temperature range of 55-80°C. The spores of some species of bacilli e.g. Bacillus stearothermophilus are extremely heat-resistant.
(e) pH:
The optimum pH for bacterial and fungal (moulds and yeasts) growth is about 7.4 and 5.6, respectively. This varies with different organisms. Strong acid or alkaline solutions are bactericidal. While pH less than 5 is avoided in case of fungi.
(f) Light:
Exposure of bacteria to sunlight in the presence of air has harmful effects. It may inhibit growth or kill the organisms. But some mould and yeast can grow equally well in the light and dark conditions. Hence, the incubators used for growing bacteria have no windows.
(g) Osmotic pressure:
Bacteria respond slowly to change in osmotic pressure, but they are plasmolysed by strong hypertonic solutions. They swell and sometimes may burst when placed in hypotonic medium. Bacterial suspensions used for test should be suspended in diluents of optimum osmotic pressure.
Mould and yeasts are more tolerant to high osmotic pressure and are often found as contaminants of unpreserved syrups, semisolid creams and ointments. When they are used in sterility testing, the inhibitory effect of strongly hypertonic solutions must be allowed for bacteria but additional sodium chloride is necessary used for moulds media.
(h) Growth inhibitors:
Many substances can inhibit the growth of bacteria and fungi. The substances that prevent the growth of bacteria and fungi without destroying them are called ‘bacteriostatic’ and ‘fungistatic’ respectively. Those substances that kill bacteria and fungi are called ‘bactericides’ and ‘fungicides’.
However, substances can be bacteriostatic at low concentrations and bactericidal at high concentrations and bacteria may die if subjected to prolonged contact with bacteriostatic concentrations. Bactericides are extensively used in injections as preservatives. Fungal inhibitors must be neutralized or ‘diluted out’ in sterility testing.
Culture Media used for Sterility Testing:
Culture media suitable for sterility testing must be capable of initiating and maintaining the vigorous growth of a small number of organisms. The organism may be aerobic and anaerobic bacteria and the lower fungi. The BP (2001) suggests the use of dual-purpose or joint media such as fluid thioglycollate and soya bean casein digest medium.
(i) Fluid Thioglycollate Medium:
Fluid thioglycollate medium is intended primarily for the culture of anaerobic bacteria but will also sustain the growth of aerobic bacteria. Resazurin is included as an oxidation- reduction indicator and agar is present to increase the viscosity.
Thus it reduces the inward diffusion of oxygen in the medium. The nutrients include sodium chloride, glucose and pancreatic digest of casein. Yeast extract is present as a growth factor and the amino acid L-cystine is included to induce the growth of certain Clostridia.
The formulation of product to be tested is suitable for the detection of anaerobes if not more than the upper third of the medium in the container, such as a 100 ml bottle, has been oxygenated. This is indicated by a green coloration of the medium. The top 10% oxygenated is very suitable for use because aerobic and anaerobic growth will be more quickly initiated under such conditions.
(ii) Soybean Casein Digest Medium:
Soybean casein digest medium intended is prepared for the growth of aerobic bacteria but it also supports the growth of fungi. Owing to the inclusion of tryptone and soya peptone, this medium is particularly supportive to fastidious aerobic bacteria that grow slowly in fluid thioglycollate medium because of the low oxidation-reduction potential. In addition, it gives good results in the membrane filtration method of sterility testing.
Sterility of the Media:
The media for sterility test must be sterile in order to eliminate the possibility of false positives results obtained from contaminated media. This assurance is obtained by incubating portions of the media at required temperature for 14 days. Media intended for detection of bacteria are incubated at 30-35°C and for fungi at 20-25°C. Sterility is confirmed by the absence of microbial growth.
The batch passes the test for sterility if there is no sign of growth in any of the test media. When microbial growth is present, the sample fails the sterility test. In such a situation the media showing growth are kept to one side and the identity of the contaminants should be determined. In such cases, the test may be repeated.
Methods for testing the sterility as defined in BP (2001) that ‘the test may be carried out using the technique of membrane filtration or by direct inoculation of the culture media with the product being examined. Appropriate negative controls are included in either case using preparation known to be ‘sterile’.
The negative control may be an ampoule or ampoules of sterile media. However, the negative control may be samples of the product being tested e.g. antibiotic powder which has been terminally sterilized. The negative control is of assistance false positives and provides a check on operator technique.
Membrane Filtration:
Membrane filtration is used for aqueous preparations, alcoholic or oily preparations, preparations miscible with or soluble in aqueous or oily solvents which do not posses antimicrobial activity under the conditions of the test. Membrane filters having a normal pore size of not greater than 0.45 ¼m proved and effective in retaining microorganisms. The filtration assembly and membrane filters are first sterilized by appropriate means.
They should be so designed that solutions to be examined are introduced and filtered under aseptic conditions. After filtration, the membrane filter can be removed intact (or divided into two) and aseptically transferred to one (or two) containers of appropriate culture medium. The identity of the microorganisms isolated implies contamination during the performance of the test for sterility i.e. false positive.
No evidence of microbial growth on subsequent retest indicates the product complies with the test for sterility and is safe. Microbial growth in the repeat test indicates that the product examined does not comply with the test for sterility. A false positive might be due to the growth of Staphylococcus epidermis, which was killed due to the preservative used in the product.
Antibiotic Assay:
Microbial growth inhibition under standard conditions may be utilized for demonstrating the therapeutic efficacy of antibiotics. Any subtle change in the antibiotic molecule which may not be detected by chemical methods will be revealed by a change in the antimicrobial activity.
Therefore, assays are very useful for resolving doubts regarding possible change in potency of antibiotics and their preparations. These are based on comparison of the inhibition of growth of microorganisms by measured concentration of the antibiotics (to be examined) with that produced by known concentrations of a standard preparation of known activity.
There are two methods usually employed for antibiotic assay:
i. Cup-plate (or cylinder-plate) method
ii. turbidimetric (or tube assay) method.
(i) Cup-plate Method:
In this method, active components of antibiotic diffuse, during incubation at optimum temperature in an appropriate medium which is lawned with a sensitive organism. The medium is gelled by agar where its chain-like molecules provide a framework in which water forms a continued capillary network through which antibiotic components diffuse.
When the antibiotic concentration is sufficiently high, microbial growth is inhibited. But at low concentrations, microbial growth is occurs. The effect of antibiotic on the microbial growth is evidenced by a zone of inhibition forming of clearing area. Most antibiotics show linear relationship between the diameter or area of this zone of inhibition and logarithm of the antibiotic concentrations.
The more effective the antibiotic, the larger is the zone of inhibition. The antibiotic solutions are paced on wells made/cut into the media, surface, or saturated on paper discs placed onto the agar.
It is useful in determining antibiotic potencies in finished pharmaceutical preparations, their intermediates or raw materials, sensitivity of pathogens in clinical material to various antibiotics, or the concentration profile of an administrated antibiotic in body fluid.
(ii) The Turbidimetric Method:
This method is less popular than agar diffusion method. It is considered to resemble more closely to the clinical situation. In the clinical situation, growth of an organism in a liquid culture is directly challenged by antibiotic treatment.
Its major use is for those antibiotics (such as gramicidin), which are poorly soluble in water and have to be prepared in another solvent such as ethanol. A nutrient broth is inoculated with an organism sensitive to the test antibiotic and dispersed into tubes containing standard antibiotic solutions.
Incubation permits the organism to grow quickly as observed by turbidity, which is inversely proportional to the logarithm of antibiotic concentration inhibiting the growth.
II. Microbial Limit Tests (MLT):
Microbial quality of non-sterile pharmaceutical products can be controlled by the adopting of one or two types of standard. The first is a limit of the total numbers of viable organisms in a given weight or volume of liquid. It is known as total viable count (TVC) and another is the total exclusion of specific pathogens.
In both the United States Pharmacopoeia (USP), European Pharmacopoeia (EP), and total viable count (TVCs) are used for different grades of organisms such as aerobic bacteria, yeast/molds or members of Enterobacteriaceae.
In case of second type, the given weight or volume of sample is tested for potentially hazardous organisms pathogenic to human or others. They may be common contaminants of raw materials or they may be indicative of the quality of the manufacturing process.
(i) Total Aerobic Microbial Count:
Is determined by microbial count in the substance being examined by using membrane filtration techniques, plate count method, multiple-tube or serial dilution method.
(ii) Tests for Specified Microorganisms:
Is carried out on the basis of detection of microbial limit test (MLT) against four bacteria namely, Escherichia coli. Salmonella typhoides, Pseudomonas aeruginosa and Staphylococcus aureus in pharmaceutical raw materials and finished products.
These organisms are recommended in the United States Pharmacopoeia (USP) (1995), European Pharmacopoeia (EP) (1997) and British Pharmacopoeia (BP) (1999). Detection of microbial limit tests is mainly applied to raw materials of natural or biological origin such as starch, gums, gelatin, etc. Tests for specified microorganisms are described in Indian Pharmacopoeia.
(a) E. coli:
It is a bacterium which is commonly encountered in the gastrointestinal tract of mammals, and detectable in their faeces. Some strains of E. coli exist harmless commensals in the intestine, while some are pathogenic and produce enterotoxins responsible for diarrhoeal disease.
Exclusion of E. coli from pharmaceutical materials is essential in order to avoid the risk of infection. Materials of natural origin are frequently in carried out limit test for E. coli because of its vulnerability to contamination.
(b) Salmonella:
Salmonella is also an inhabitant of mammalian intestine. Its species are also subjected to microbial limit tests. Its pathogenic potential is substantially greater than that of E. coli since harmless strains of Salmonella are less frequently encountered. Some strains can initiate infections from the ingestion of very small numbers of cells.
(c) Staphylococcus aureus:
It is inhabits in the human skin and nose without any apparent ill effects. However, some strains exhibit a marked pathogenic potential and cause serious infections. They may originate on the skin but progress to other sites (anatomical), possibly with life-threatening impacts.
The presence of S. aureus as contaminant may be an indicator of poor quality product which reveals the non-sterile conditions during product manufacturing. Generally it sheds through the skin of personnel working in the plant.
(d) Pseudomonas aeruginosa:
It has gained much attention for several reasons. It is a potential pathogen capable of causing infection at most vulnerable sites in healthy person e.g., eyes. It is regarded as an opportunist pathogen which may cause infection in many regions of the body with underlying disease or impaired immunity.
As far as its pathogenic potential is concerned, the organism would be a potential problem as contaminant of pharmaceutical material because the isolates are found to be commonly resistant to preservatives. This bacterium is a fast growing with low nutritional requirements.
It can use a wide variety of organic material (at low concentrations) as nutrients and achieve relatively high cell densities. Water is one of natural habitats of Pseudomonas aeruginosa, and stored water is more likely to be contaminated than freshly purified water. Testing of these four bacteria is described in Practical Microbiology by Dubey and Maheshwari (2006).
Pyrogen:
Pyrogens or endotoxins are such substances that produce fever. The terms pyrogen and endotoxin are synonym of each other. All endotoxins are pyrogens but all pyrogens are not endotoxins. Some pyrogens are chemical substances also. Water is potentially the greatest source of pyrogens in parenteral drugs.
Untreated water is contaminated with pyrogen therefore must be removed before using the water in parenteral products. The injection of distilled water may produce a rise in body temperature if it contains pyrogens. Water free from this effect is described as ‘apyrogenic’. Freshly prepared parenteral products must not be contaminated with organisms that could produce pyrogens.
The products must be prepared in conditions that reduce microbial contamination because bacteria contaminating aqueous solutions can release endotoxins. Contaminated solutions will become more pyrogenic with the passage of time. Therefore, the products must be sterilized quickly after preparation.
It is a well known fact that pyrogenic substances cause a rise in body temperature following intravenous administration. There are many biological and chemical pyrogens, but the most significant pyrogen encountered in the routine production parenteral and medical devices is Gram- negative bacterial endotoxin.
Bacterial endotoxin consists of lipopolysaccharide (LPS) molecules which forms a part of membrane or cell wall of Gram-negative bacteria. The toxin is shed into the surrounding medium in copious quantity following bacterial lysis due to death or antibacterial action such as heat sterilization or other chemical/physical processes.
Therefore, endotoxin is associated with the cell wall of living bacteria or found in freely. The molecular structure of Salmonella lipopolysaccharide consists of hydrophobic lipid-A attached to a core region which includes three 2-ket-3-de-oxyoctonate (KDO) molecules (Fig. 19.7).
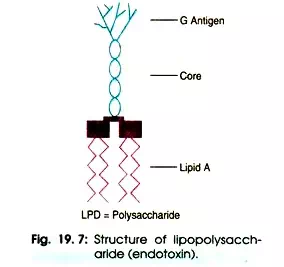
The polysaccharide chain is highly variable amongst different bacteria. The size of endotoxin is 10 kDa but it can form large aggregates up to 1000 kDa. After exposure to endotoxins humans produces antibodies directed at the polysaccharide chain.
The molecule is amphipathic, aggregate in aqueous solution. Some toxin is heat stable, so it is essential that endotoxin contamination is avoided during manufacturing of parenterals and medical devices because it cannot further be removed by autoclaving.
The only Gram-positive bacteria that produces endotoxin is Listeria monocytogenes, but such toxins are more accurately described as ‘endotoxin-like’ just to differentiate from the toxins found in the cell wall of Gram-negative bacteria.
Bacterial endotoxin causes several physiological effects following intravenous injection such as activation of the cytokine system, endothelial cell damage, drop in blood pressure and intravascular coagulation.
When body is subjected to massive doses of toxin, it ultimately causes death due to septic shock. In most Pharma companies, their microbiology laboratory is involved with the quality control of parenterals and medical devices as the workload of bacterial endotoxin test (BET).
The presence of endotoxins in the blood is called ‘endo-toxemia’. If the immune response is severely pronounced, it can lead to septic shock. LPS binds to the lipid binding protein in the human blood serum which transfers it to CD 14 on the cell membrane.
In turn it transfers to another non- anchored protein, MD2, which associates with Toll-like receptor-4 (TLR4). CD 14 and TLR4 are present in different types of cells of immune system such as macrophages and dendritic cells. It triggers the signalling cascade for macrophage/endothelial cells to secrete the pro-inflammatory cytokines and nitric oxide that lead to ‘endotoxic shock’.
During 1980s, rabbit pyrogen test was replaced by Limulus amoebocyte lysate (LAL) test emerged as alternative method. Use of the rabbit test has declined rapidly in the 1990s, but still this test has been carried out on to older products which have not been converted to the LAL Test. According to FDA guidelines (1983), endotoxin limits was standardized for all drugs except intrathecals from 2.5 EU Kg-1 to 5.0 EU Kg-1.
1920’s Seibert proved fevers from IV products were induced by heat-stable, filterable substances called pyrogens and then he chose the rabbit as the animal model. But Rabbits test may cause false positives. Moreover, it needs to inject three rabbits per test.
Rabbit Pyrogen Test:
The rabbit in vivo pyrogen test was a historical milestone for examination of injectable medicines. Since the 1940’s all parenteral drugs must be tested to prove that they are pyrogen free when injected in rabbits. A drag sample is injected into three rabbits. The rabbits must be individually held in a fixed position for a number of hours in a narrow cage.
A temperature probe is placed in the rectum of individual rabbit to record an increase in body temperature. An increase in their body temperature gives an indication that the drug has pyrogen contamination.
The function of an increased body temperature is still quite unclear. However, it is thought that the replication of viruses and bacteria is decreased by fever, antigen processing is speeded up, and the specific immune response is intensified.
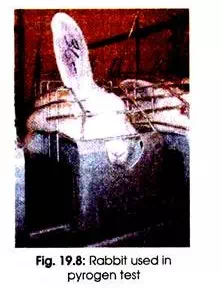
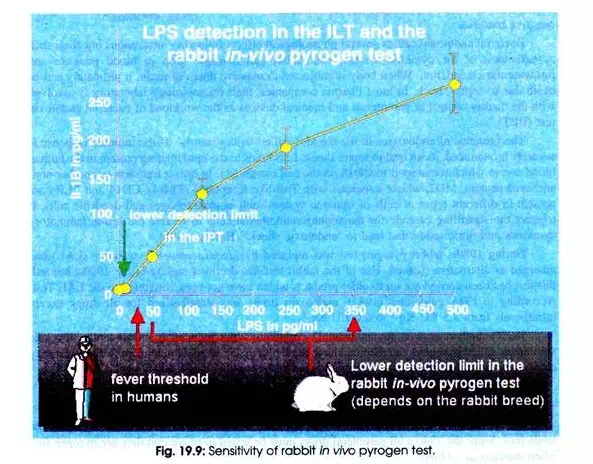
The rabbit pyrogen test required by the authorities is very reliable; otherwise many more complications would be expected to occur after the injection of tested drugs. But the rabbit pyrogen test has its limitation. The sensitivity of the test quietly depends on the type of rabbits used.
The sensitivity of this test lies between 50 and 350 pico gram (pg) LPS/ml (Fig. 19.9). There arises a question whether a person or a rabbit will respond with the same sensitivity to a given concentration of a pyrogenic substance. Unfortunately, it does not. Human beings show a fever response at 25pg LPS/ml. In practice, it is balanced out by applying a higher dose to the test rabbit as ever will be given to patients.
In Vitro Pyrogen Test (IPT):
Nevertheless, the in vitro pyrogen test provides a decisive advantage. Its detection limit for LPS lies at 10pg LPS/ml. The new IPT is relevant for the 3R- idea. Animal protection concerns are followed up in that IPT procedures will be adapted for animal blood, to allow for example the examination of air quality in animal stalls, and thus the study and improvement of animal husbandry.
In IPT a very small volume (100 µl) of heparinized full blood is taken by a standard procedure, diluted with physiological salt solution (1 ml), and the sample (100µl) of a pyrogenic contamination to be tested is added. The same response occurs in vitro as would occur in a living organism. Cytokines are excreted by macrophages, amongst others interleukin 1-beta.
After incubation of reaction mixture for overnight at body temperature, the concentration of interleukin 1-beta in the supernatant is measured with an ELISA (Fig. 10.10). Interleukin 1-beta is excreted in the same way as other cytokines by macrophages, which come into contact with particular extrinsic substances i.e. pyrogens for example.
The cytokines on their part then trigger an immune response as a defensive reaction. Interleukin 1-beta can only be found in human blood in connection with such an inflammatory reaction. The effects of Interleukin 1-beta in the body are manifold.
The acute-phase reaction is activated in the liver which means that proteins of the phase of acute inflammation e.g. C- reactive protein and mannan binding lectins are produced. In the hypothalamus the target temperature in the temperature regulation centre is increased that results in rise in body temperature.

Endotoxin (Limulus Amebocyte Lysate (LAL)) Test:
The LAL test was developed during the mid-1960s by Levin and Bang (1964). The LAL test is used to detect endotoxins associated with Gram-negative bacteria.
The lysate, prepared from the circulating blood cells (amebocytes) of the horseshoe crab (Limulus polyphemus) (Fig. 19.11) is more sensitive to the detection of endotoxin than the rabbit pyrogen test. The test is based on Gram- negative bacterial infection of Limulus polyphemus resulted in fatal intravascular coagulation.
Levin and Bang (1964) demonstrated that clotting was due to the action between endotoxin and a clottable protein in the circulating amoebocytes of Limulus. The suitable anticoagulant for Limulus blood was prepared from lysate from washed amoebocytes which proved an extremely sensitive indicator for the presence of endotoxin. Solum (1973) and Young et al. (1972) purified and characterized the clottable protein (enzyme) from LAL which is reactive to that of endotoxin.

The LAL test utilizes the basic immune response of the horseshoe crab to Gram-negative bacterial invasion. The horseshoe crab amebocyte constitutes several proteins, factors, co-factors and ions that interact to initiate coagulation. Endotoxins of Gram-negative bacteria catalyze the activation of proenzyme in Limulus amebocyte lysate.
Its initial rate of activation is inferred by the concentration of endotoxin present. The activated enzyme (coagulase) breaks specific bonds in clotting protein (coagulogen) present in Limulus amebocyte lysate. Once protein is hydrolysed, the resultant coagulin self-associates and forms a gelatinous clot. There are three methods namely gel clot, turbidimetric, and chromogenic methods for carrying out the LAL test.
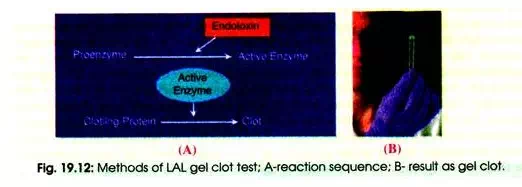
(i) Gel Clot Assay Method:
The gel clot assay was the original LAL method and relies upon the operator to distinguish the formation of the gel clot in the reaction tubes. It is a qualitative or semi-quantitative test that is used to screen for the presence of endotoxins. The specimen is incubated with LAL of a known sensitivity (19.12 A). Formation of a gel clot is positive result for the presence of endotoxin in the sample (B).
If no clot forms, this is interpreted as the sample being endotoxin free. The results are from the subjective interpretation of the clot formation. The gel clot method is based on the fact that LAL clots in the presence of endotoxin. It is an in vitro test for bacterial endotoxins. It is quick, simple to perform and sensitive to low endotoxin concentrations.
(ii) Turbidimetric Method:
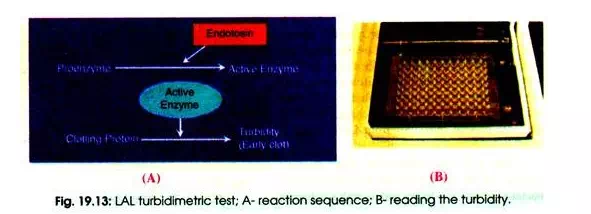
Some products are of a colour that would interfere with this form of testing, and so the turbidimetric method can be used to avoid any such interference. In this case a different lysate is used and the reaction with endotoxin results in the solution becoming turbid, thus allowing quantitation of endotoxin content without relying on the colour present.
The turbidimetric method which is a kinetic and quantitative LAL method utilizes more sophisticated kinetic spectrophotometers to monitor turbidity by which one can determine endotoxin content. The specimen is incubated with LAL and either the rate of increase in turbidity or the time taken to reach a particular turbidity is measured spectrophotometrically and compared to a standard curve (Fig. 19.13 A, B).
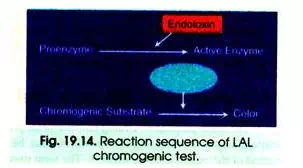
(iii) Chromogenic Method:
The third method is a chromogenic method which employs a synthetic chromogenic substrate. In the presence of LAL and endotoxin the chromogenic substrate produces a yellow colour that is linearly related to the concentration of endotoxin. (Fig. 19.14).
Most common problem encountered by all users of the LAL test is the frequent interference of with the assay from the test compound. Guilfoyle and Munson (1982) demonstrated that out 587 compounds tested, 11% compounds interfered with LAL assay to some extent.
Control Standard Endotoxin (CSE):
The CSE has a predetermined amount of endotoxin, as described in the ‘certificate of analysis’ (COA), which has standardized with the U.S. ‘reference endotoxin. The lyophilized endotoxin (CSE) is prepared according to the instruction mentioned in the COA.
Determination of Endotoxin Limits:
It is necessary to calculate the endotoxin limit for the preparation or item before the testing of final products. If there is no predetermined limit available from pharmacopoeia, it must be calculated from the maximum human dose.
Different regulatory bodies named endotoxin limit with other names also such as maximum allowable endotoxin concentration (MAEC), endotoxin release limit (ERL) or endotoxin limit concentration (ELC).
If there is no monograph available, the endotoxin limit may be determined from the formula as given below:
Endotoxin limit = K/M
Where K = constant equal to 5 EU (endotoxin unit) per kg of body weight
M = maximum human dose administrated per kg/hour.
Maximum Valid Dilution (MVD) or Minimum Valid Concentration (MVC):
The maximum valid dilution (MVD) or minimum valid concentration (MVC) is calculated figures to indicate the degree to which a product may be diluted to overcome interference. Sometimes, the effect of the dilution exceeds the ability (LAL test method being used) to detect endogenous endotoxin in the original preparation.
The term MVD is normally applied to preparations already in a liquid form where the dose is administrated per milliliter e.g. a single 2ml injection, the endotoxin limit is expressed as EUml-1.The term MVC is applied to those preparations where the endotoxin limit is expressed as EU mg-1 and dose is expressed as mg/kg of body weight.
According to the manufacturer’s instructions, accurate volume of sample or standards prepared from the CSE (typically 100µl) is pipetted into (10x75mm) depyrogenated glass tubes.
The kit normally describes the correct dilution schemes. It usually comprises two-fold dilutions of an endotoxin standard, diluted test sample and LAL reagent water to serve as a negative control. After addition of 100 µl reconstituted lysate, all the tubes are kept on 37°C for a period of 1 h incubation period.
After incubation, each tube in the gel-clot method is interpreted as either positive or negative. A positive test is defined as the formation of a firm gel capable of maintaining its integrity when the test tube is inverted at 180°. A negative test is characterized by the absence of gel due to formation of a viscous mass which does not hold after inverting the tube.
III. Preservative Efficacy Test (PET):
The main purpose of adding antimicrobial preservatives is to prevent the adverse effects from contamination by microorganisms which may be introduced inadvertently during or subsequent period of the manufacturing process. The term preservative means any chemical agent that protects a product from degradation or change which occurs due to microbial contamination.
However, this can be misleading since it might be thought that preservative merely prevent growth of microorganisms, but do not kill them. In many cases, the concentrations of preservative used in product formulations are designed to give a rapid killing of invading microorganisms.
Interestingly, preservatives are used in combination, as a broader spectrum of antimicrobial cover or enhanced activity, and other chemicals which themselves have little or no antimicrobial activity. Certain chemicals are combined with a preservative in order to enhance its activity e.g. EDTA (ethylene diamine tetra-acetic acid).
(i) Factors Affecting Activity of Preservatives:
There are several factors which will influence activity of preservative as given below:
(a) pH:
Some preservatives such as sorbic acid and benzoic acid are affected by pH because pH influences the ionic state of the compound. In order to enter in a microbial cell and to show antimicrobial effect, weak organic acids must be unionized which may occur under conditions of low pH. At high pH, the biocidal activity of these weak acids is much reduced. During storage, it is possible the pH may be increased but the concentration of the preservative remains unchanged.
(b) Presence of non-aqueous phase:
Some cosmetics and pharma products in the form of creams and emulsions have non-aqueous phases. It does not usually require preservation since microorganisms may grow almost exclusively in the aqueous phase of the formulation or at the interface.
However, depending upon the partition coefficient of the preservative, some of the biocidal action within the aqueous phase may be lost if the compound is highly soluble in the oil phase. Partition can be an extended process encouraged by certain storage conditions.
(c) Adsorption to suspended solids:
Suspended solids contained within suspensions may adsorb preservatives, thus reducing the concentration of preservatives in the aqueous phase to sub- effective levels.
(d) Adsorption onto plastic packaging:
Glass containers do not usually represent a problem. But to increasing use of plastics, considerations must be given to the choice of preservation. Some compounds may be absorbed into the plastics and subsequently evaporate out from the container. Test of preservative is described in Practical Microbiology by Dubey and Maheshwari (2007).
(ii) Limitations of Preservative Efficacy Test:
Despite its apparent simplicity, the preservative efficacy tests pose a number of practical problems. Many preservatives in aqueous solution exhibit first-order kill kinetics.
Log percentage survivors are plotted as a function of time (to give to plot a kill curve or survivor plot). The graph may show the presence of shoulders (lag) or tails rather than the expected linear relationship. There are number of reasons for non-linear plots including the presence of resistant cells within a population.
Sometimes it is due to lysis of dead cell debris which affords protection to those which are still alive. Non-linear kill curves can make interpretation of preservative efficacy data complicated, particularly when only one or two time points are used for sampling.