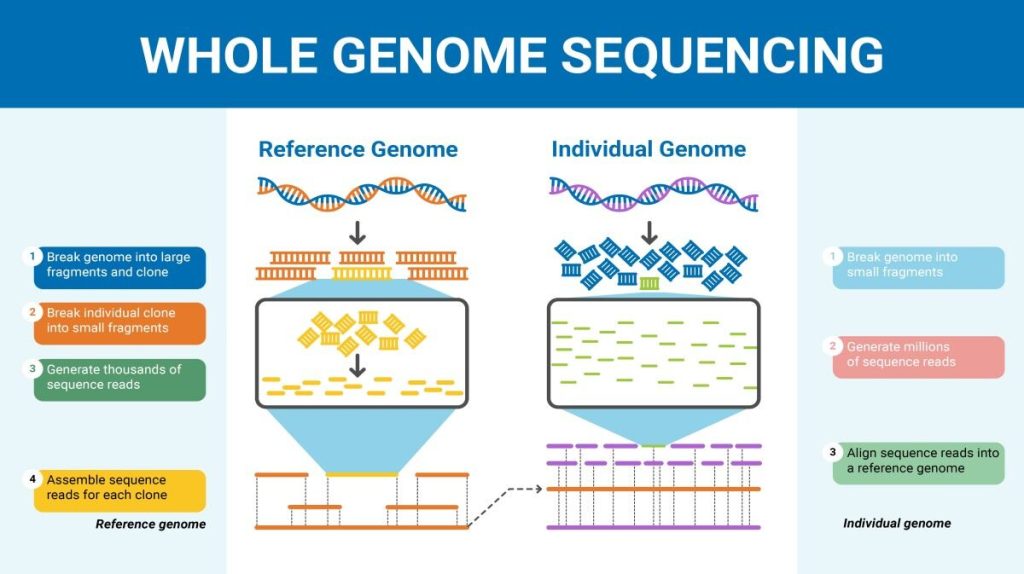
Although there have been significant advances in the medical sciences in recent years, doctors are still confounded by many diseases and researchers are using whole genome sequencing to get to the bottom of the problem. Whole genome sequencing is a process that determines the DNA sequence of an entire genome. Whole genome sequencing is a brute-force approach to problem solving when there is a genetic basis at the core of a disease. Several laboratories now provide services to sequence, analyze, and interpret entire genomes.
In 2010, whole genome sequencing was used to save a young boy whose intestines had multiple mysterious abscesses. The child had several colon operations with no relief. Finally, a whole genome sequence revealed a defect in a pathway that controls apoptosis (programmed cell death). A bone marrow transplant was used to overcome this genetic disorder, leading to a cure for the boy. He was the first person to be successfully diagnosed using whole genome sequencing.
The first genomes to be sequenced, such as those belonging to viruses, bacteria, and yeast, were smaller in terms of the number of nucleotides than the genomes of multicellular organisms. The genomes of other model organisms, such as the mouse (Mus musculus), the fruit fly (Drosophila melanogaster), and the nematode (Caenorhabditis elegans) are now known. A great deal of basic research is performed in model organisms because the information can be applied to other organisms. A model organism is a species that is studied as a model to understand the biological processes in other species that can be represented by the model organism. For example, fruit flies are able to metabolize alcohol like humans, so the genes affecting sensitivity to alcohol have been studied in fruit flies in an effort to understand the variation in sensitivity to alcohol in humans. Having entire genomes sequenced helps with the research efforts in these model organisms (Figure 10.12).

Figure 10.12 Much basic research is done with model organisms, such as the mouse, Mus musculus; the fruit fly, Drosophila melanogaster; the nematode Caenorhabditis elegans; the yeast Saccharomyces cerevisiae; and the common weed, Arabidopsis thaliana. (credit “mouse”: modification of work by Florean Fortescue; credit “nematodes”: modification of work by “snickclunk”/Flickr; credit “common weed”: modification of work by Peggy Greb, USDA; scale-bar data from Matt Russell)
The first human genome sequence was published in 2003. The number of whole genomes that have been sequenced steadily increases and now includes hundreds of species and thousands of individual human genomes.
Applying Genomics
The introduction of DNA sequencing and whole genome sequencing projects, particularly the Human Genome Project, has expanded the applicability of DNA sequence information. Genomics is now being used in a wide variety of fields, such as metagenomics, pharmacogenomics, and mitochondrial genomics. The most commonly known application of genomics is to understand and find cures for diseases.
Predicting Disease Risk at the Individual Level
Predicting the risk of disease involves screening and identifying currently healthy individuals by genome analysis at the individual level. Intervention with lifestyle changes and drugs can be recommended before disease onset. However, this approach is most applicable when the problem arises from a single gene mutation. Such defects only account for about 5 percent of diseases found in developed countries. Most of the common diseases, such as heart disease, are multifactorial or polygenic, which refers to a phenotypic characteristic that is determined by two or more genes, and also environmental factors such as diet. In April 2010, scientists at Stanford University published the genome analysis of a healthy individual (Stephen Quake, a scientist at Stanford University, who had his genome sequenced); the analysis predicted his propensity to acquire various diseases. A risk assessment was done to analyze Quake’s percentage of risk for 55 different medical conditions. A rare genetic mutation was found that showed him to be at risk for sudden heart attack. He was also predicted to have a 23 percent risk of developing prostate cancer and a 1.4 percent risk of developing Alzheimer’s disease. The scientists used databases and several publications to analyze the genomic data. Even though genomic sequencing is becoming more affordable and analytical tools are becoming more reliable, ethical issues surrounding genomic analysis at a population level remain to be addressed. For example, could such data be legitimately used to charge more or less for insurance or to affect credit ratings?
Genome-wide Association Studies
Since 2005, it has been possible to conduct a type of study called a genome-wide association study, or GWAS. A GWAS is a method that identifies differences between individuals in single nucleotide polymorphisms (SNPs) that may be involved in causing diseases. The method is particularly suited to diseases that may be affected by one or many genetic changes throughout the genome. It is very difficult to identify the genes involved in such a disease using family history information. The GWAS method relies on a genetic database that has been in development since 2002 called the International HapMap Project. The HapMap Project sequenced the genomes of several hundred individuals from around the world and identified groups of SNPs. The groups include SNPs that are located near to each other on chromosomes so they tend to stay together through recombination. The fact that the group stays together means that identifying one marker SNP is all that is needed to identify all the SNPs in the group. There are several million SNPs identified, but identifying them in other individuals who have not had their complete genome sequenced is much easier because only the marker SNPs need to be identified.
In a common design for a GWAS, two groups of individuals are chosen; one group has the disease, and the other group does not. The individuals in each group are matched in other characteristics to reduce the effect of confounding variables causing differences between the two groups. For example, the genotypes may differ because the two groups are mostly taken from different parts of the world. Once the individuals are chosen, and typically their numbers are a thousand or more for the study to work, samples of their DNA are obtained. The DNA is analyzed using automated systems to identify large differences in the percentage of particular SNPs between the two groups. Often the study examines a million or more SNPs in the DNA. The results of GWAS can be used in two ways: the genetic differences may be used as markers for susceptibility to the disease in undiagnosed individuals, and the particular genes identified can be targets for research into the molecular pathway of the disease and potential therapies. An offshoot of the discovery of gene associations with disease has been the formation of companies that provide so-called “personal genomics” that will identify risk levels for various diseases based on an individual’s SNP complement. The science behind these services is controversial.
Because GWAS looks for associations between genes and disease, these studies provide data for other research into causes, rather than answering specific questions themselves. An association between a gene difference and a disease does not necessarily mean there is a cause-and-effect relationship. However, some studies have provided useful information about the genetic causes of diseases. For example, three different studies in 2005 identified a gene for a protein involved in regulating inflammation in the body that is associated with a disease-causing blindness called age-related macular degeneration. This opened up new possibilities for research into the cause of this disease. A large number of genes have been identified to be associated with Crohn’s disease using GWAS, and some of these have suggested new hypothetical mechanisms for the cause of the disease.
Comments are closed