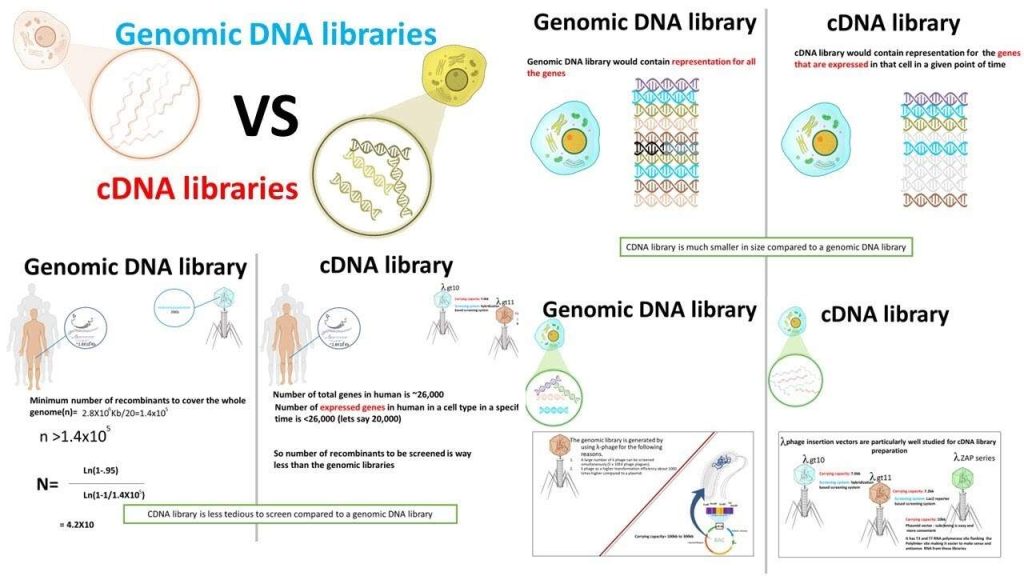
Isolation of corresponding genetic information
Instead of synthesizing a desired gene, can we used the amino acid information to directly isolate the corresponding genetic information?
· There are two general sources of genetic information:
1. Genomic DNA
2. mRNA
If we are considering genomic DNA from eukaryotes, then there are a couple of things to consider:
1. The coding region for a gene of interest may be interrupted by one or more intron regions, and thus the complete coding region could be quite long.
2. To a first approximation, it does not matter which tissue we use to isolate the genomic information, i.e. the genomic content is the same in all tissues.
If we are considering mRNA from eukaryotes, we may realize the following advantages:
1. Introns will be spliced out and the mRNA will contain a contiguous coding region.
2. Tissue specific expression of the protein of interest may allow us to isolate appropriate mRNA at enhanced levels, i.e. in tissues where the protein is expressed the mRNA levels are considerably higher than the corresponding genomic levels (there are many more molecules of mRNA than copies of the gene).
Libraries
A “library” is a convenient storage mechanism of genetic information.
· They are typically either “genomic” or “cDNA” (i.e. mRNA in DNA form) genetic information.
· Deduced genetic sequences from corresponding polypeptide information can be used to identify specific genetic information within a library.
cDNA library construction
“Reverse transcription” is a mechanism whereby genetic information contained in mRNA is converted back into a double stranded DNA form.
The enzyme responsible for this is an RNA dependent DNA polymerase called reverse transcriptase.
· Reverse transcriptases have traditionally been isolated from viruses whose genome is actually in an RNA form and must be converted to duplex DNA.
· These viruses typically carry a functional reverse transcriptase along with their mRNA genetic component when they infect cells.
· One of the most common commercially available reverse transcriptases is Moloney murine leukemia virus (MMLV).
· This RNA dependent DNA polymerase (as will all polymerases) add nucleotides to a nacent polynucleotide in the 5′ to 3′ direction using RNA as the template. It does not contain any 3′->5′ exonuclease (proofreading) activity.
MMLV will use mRNA as a template, but requires a primer (it can extend a DNA primer but cannot synthesize one).
· One of the really neat things about eukaryotic mRNA’s is the presence of the 3′ poly A tracks.
Therefore we can use poly dT as a single primer for a variety of different eukaryotic mRNA’s.
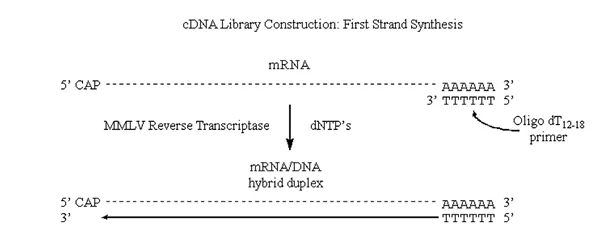
Figure 3.6.1: First strand synthesis
Note that we have produced complementary DNA (or cDNA) to the original mRNA strand.
If we can introduce “nicks” into the RNA half of this DNA/RNA duplex then the situation would be very similar to that observed in “lagging strand” synthesis of prokaryotic genomic DNA.
· Nicks in the RNA half of the molecule can be introduced via the action of the enzyme RNAse H.
· This enzyme exhibits endonucleolytic cleavage of the RNA moiety of RNA/DNA hybrids, as well as 5′->3′ and 3′->5′ exoribonuclease activity.
· In other words, it will nick the RNA and then proceed to digest back in both directions:
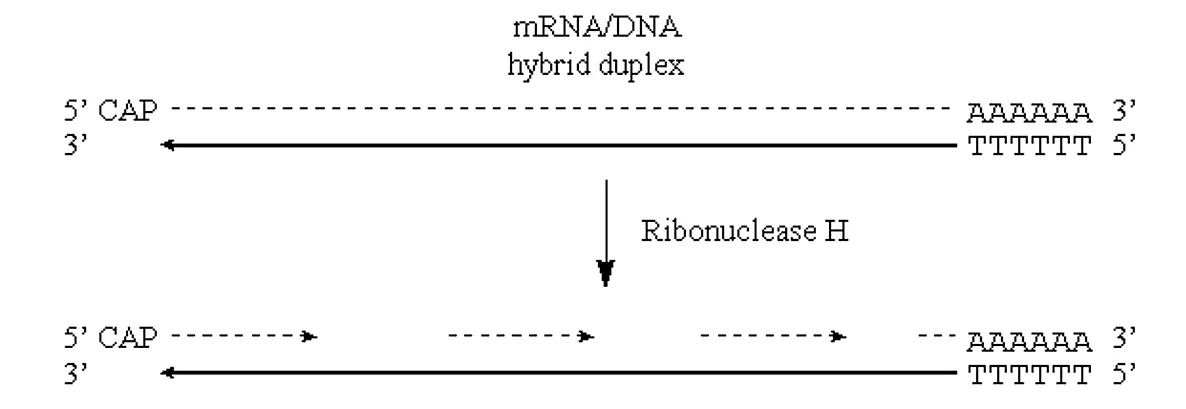
Figure 3.6.2: Nicks in mRNA
· These RNA fragments can now serve as primers for DNA synthesis by E. coli Pol I. This enzyme will also translate the “nicks” to effectively remove the RNA primers:
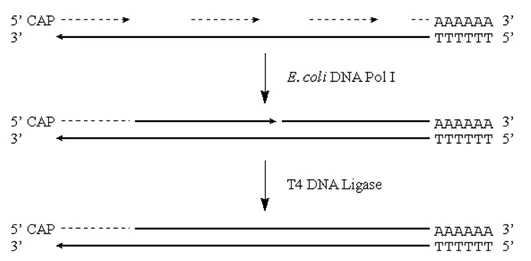
Figure 3.6.3: DNA synthesis
Note that we will potentially have either a residual 5′ RNA cap region, or a gap at the 5′ end of the original mRNA strand.
Insertion of cDNA into plasmid.
To complete our construction of a useful cDNA library we need a way to maintain and propagate our cDNA.
· We can accomplish this by inserting the cDNA into an appropriate plasmid.
· There are two classical ways of accomplishing this feat:
1. Homopolymeric tailing
2. Linker addition
Homopolymeric tailing
Terminal transferase is an unusual DNA polymerase found only in a type of eukaryotic cell called a prelymphocyte.
· In the presence of a divalent cation the enzyme catalyzes the addition of dNTP’s to the 3′-hydroxyl termini of DNA.
· When the nucleotide to be added is a purine, Mg2+ is the cation used.
· When the nucleotide to be added is a pyrimidine, Co2+ is used.
· Depending on the reaction conditions, anywhere from three to several thousand bases will be added.
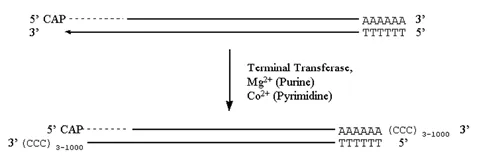
Figure 3.6.4: Terminal transferase activity
How can we insert this into a plasmid?
· If we cut our plasmid and also treat it with terminal transferase, except now we add the complementary base to the one we added to our cDNA, we can anneal and ligate the cDNA into the plasmid.
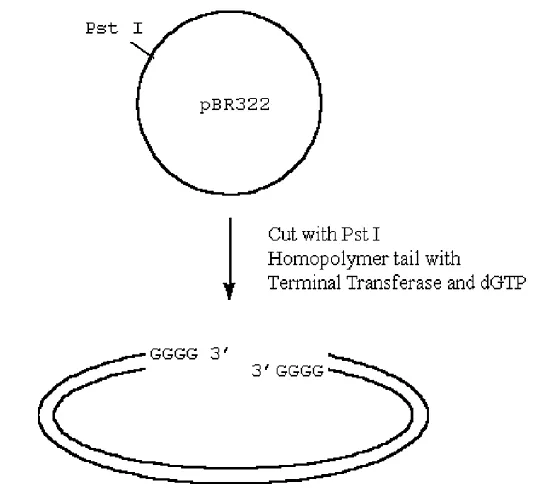
Figure 3.6.5: Ligating cDNA into the plasmid
· The utility of inserting the C-tailed cDNA insert into a G-tailed Pst I site in the vector is as follows:
1. The Pst I recognition sequence and cleavage site is
5′ C T G C A G 3′
3′ G A C G T C 5′
2. Cleavage of this site by Pst I, followed by G-tailing will produce
5′ C T G C A (G)n G 3′
3′ G (G)n A C G T C 5′
Thus, the Pst I recognition cleavage site is regenerated and the C-tailed cDNA insert can be excised again from the library vector by cleaving with Pst I.
Linkers
An alternate method to insert cDNA fragments into a library vector is through the addition of “linkers”.
· Linkers are short oligonucleotides (~18 to 24 mers) which are typically palindromic and contain a single or repeated restriction endonuclease recognition sequence.
· The palindromic nature allows the linker oligonucleotide to self-hybridize to form a blunt ended duplex.
· If the ends of the cDNA fragments are blunt, then the linker can be ligated to both ends to introduce useful terminal restriction sites.
The steps in linker addition are as follows:
1. Treatment of cDNA with S1 nuclease (to remove possible 5′ cap mRNA fragment remaining in cDNA duplex
2. Convert potential “ragged” ends to blunt by treatment with Pol I (will fill in 5′ overhangs and chew back 3′ overhangs)
3. Methylate cDNA at potential internal Eco RI sites by treatment with Eco RI methylase (plus S-adenosyl methionine)
4. Ligate linkers to blunt, methylated cDNA using T4 DNA ligase
5. Cut linkers with Eco RI restriction endonuclease
6. Remove linker fragments from cDNA fragments by agarose gel electrophoresis
7. Ligate cDNA to vector DNA fragment (opened up by Eco RI restriction endonuclease