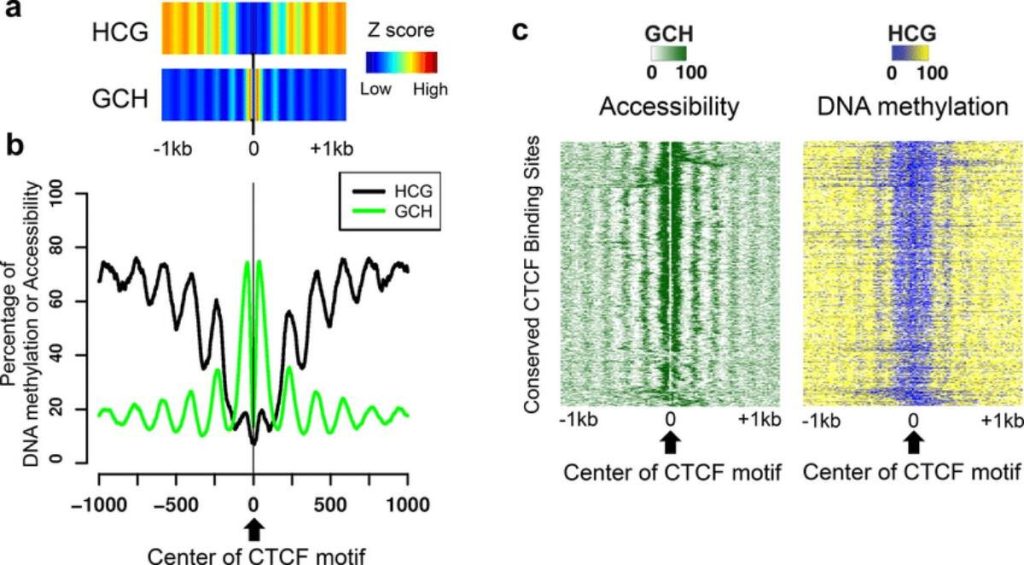
NOMe-seq doesn’t involved tiny gnomes that magically tell you all the epigenetic information of a cell. It does give you some pretty magical insight that other techniques don’t though. The real innovation of NOMe-seq is the use of a de novo GpC methyltransferase. Formaldehyde-fixed chromatin is treated with the GpC methyltransferase. Any GpC’s that are blocked by nucleosomes or other proteins will not become methylated. The cross-links are then reversed and the DNA is bisulfite treated and sequenced. The BS-seq serves two functions. First, it tells you which CpGs are methylated. Second, since GpC’s not never methylated in the mammalian genome, any that are present imply open chromatin at that location. GpC’s are also frequent enough that a high-resolution map of nucleosome occupancy is generated. Both of these data sets are generated from the same sequencing run. NOMe-seq was developed by Kelly et al., (2012). Using the technique, they found an anti-correlation of nucleosome occupancy and CpG methylation around non-promoter CTCF sites. Also, nucleosome organization at CpG island and non-CpG island promoters was found to be similar. NOMe-seq has also been used to characterize the epigenetic state of the H19 locus (Ito et al., 2013).
Posted inGenetic Engineering Updates
NOMe-seq (Nucleosome Occupancy and Methylome Sequencing)
Post navigation
Previous Post
 GENOME SEQUENCING
GENOME SEQUENCINGNext Post
GENOME VARIATIONS 