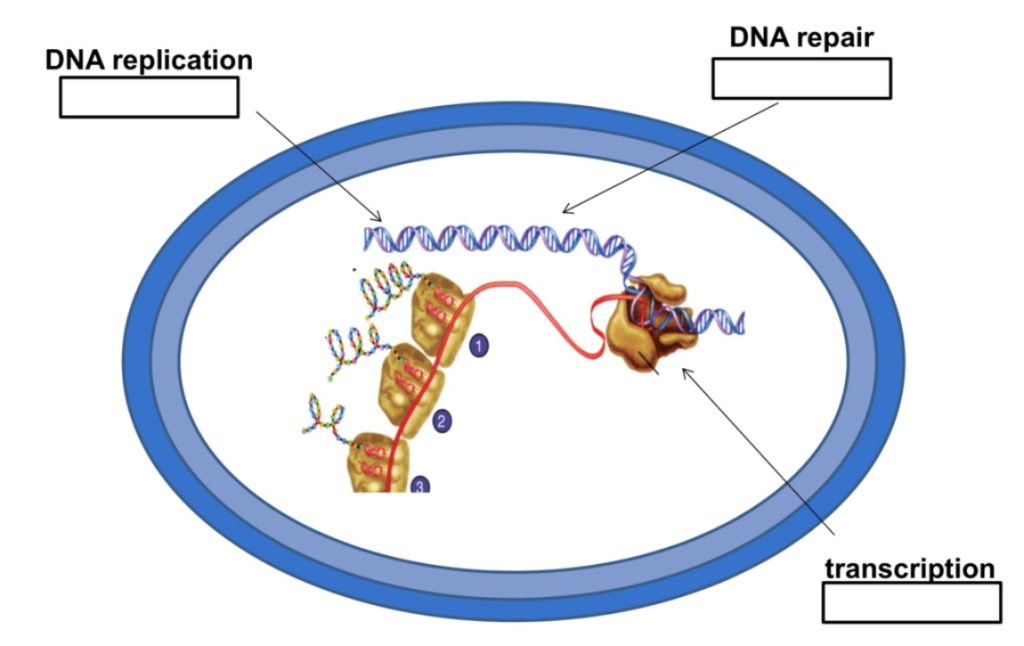
In a bacterial cell, or any kind of cell for that matter, the nucleic acids DNA and RNA are incredibly important molecules. When a cell divides, it must first replicate its DNA to give the new cell what basically amounts to its instruction manual for life. And in the daily life of a cell, transcription of DNA into RNA is a major step in the assembly line that creates proteins. It’s easy to see that if DNA or RNA synthesis are inhibited, a cell won’t be able to get anything done at all!
So, inhibiting nucleic acid synthesis sounds like a great strategy for an antibiotic. And luckily for us, the enzymes that carry out DNA and RNA synthesis are different enough between eukaryotic and prokaryotic cells that selective toxicity can be achieved. In this lesson, we’ll learn about two major classes of antibiotics that inhibit nucleic acid synthesis: rifamycins and quinolones. We’ll see how these antibiotics work, why they’re selectively toxic, and how bacteria can become resistant to them.
Rifamycins
The rifamycins are a family of antibiotics that inhibit bacterial RNA polymerase. Rifamycins work by binding to the bacterial DNA-dependent RNA polymerase, the enzyme that is responsible for transcription of DNA into RNA. The antibiotic molecule is thought to bind to the polymerase in such a way that it creates a wall that prevents the chain of RNA from elongating. Rifamycins are bactericidal antibiotics. In the presence of rifamycins, bacteria can’t transcribe any genes that they need to carry out their normal functions, so they die.
Rifamycins are broad-spectrum antibiotics, meaning they’re effective against many types of bacteria, including Gram-negative, Gram-positive, and obligate intracellular bacteria. There are two main reasons for this. First, the rifamycin molecule can penetrate well into cells and tissues. This means that, unlike some antibiotics that can’t cross certain types of bacterial cell walls, the rifamycins can almost always get in and gain access to their target enzyme. And second, the bacterial RNA polymerase is well-conserved even among very different bacteria. This means that the enzyme’s structure is similar enough that the rifamycins can bind well to their target in diverse types of bacteria.
And how do the rifamycins achieve selective toxicity? After all, our cells need RNA polymerases too! Luckily for us, rifamycins do not bind to eukaryotic RNA polymerases, so our own cells can continue to transcribe genes normally even when we are taking these antibiotics.
The best-known and most effective member of the rifamycin family is rifampin, which is also known as rifampicin. A major use of rifampin is in the treatment of mycobacterial diseases, such as tuberculosis and leprosy. Since mycobacteria are obligate intracellular bacteria, they live within host cells, where they’re protected against many antibiotics that can’t get inside. Rifamycins can penetrate well into cells and tissues, so they’re a good first choice for mycobacterial infections. However, as with any antibiotic, there are bacteria that are resistant to the rifamycins. The most common way for bacteria to become resistant to rifamycins is to acquire mutations that alter the structure of the RNA polymerase in such a way that rifamycins can’t bind to it as well.
Quinolones and Fluoroquinolones
The second major class of antibiotics that inhibit nucleic acid synthesis is the quinolones and their derivatives, the fluoroquinolones. These are synthetic antibiotics that were first developed in the 1960s. Drugs in this family, such as nalidixic acid, ciprofloxacin, and norfloxacin, work by inhibiting enzymes that are required for bacterial DNA synthesis. So, in contrast to the rifamycins, which inhibit transcription of DNA into RNA, the quinolones and fluoroquinolones inhibit DNA replication. But fortunately for us, they don’t bind to eukaryotic enzymes for DNA replication, so they’re selectively toxic.