An overview of hydrogenation and dehydrogenation
Many of the redox reactions that you will encounter when studying the central metabolic pathways are classified as hydrogenation or dehydrogenation reactions. Hydrogenation is simply the net addition of a hydrogen (H2H2) molecule to a compound, in the form of a hydride ion (H−H−, a proton plus a pair of electrons) and a proton. Hydrogenation corresponds to reduction. Dehydrogenation reactions are the reverse process: loss of a hydride and a proton. Dehydrogenation corresponds to oxidation.
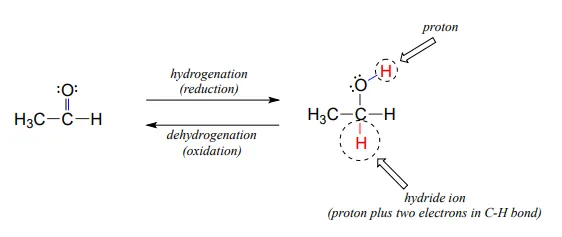
Hydrogenation and dehydrogenation reactions can also be called hydride transfer reactions, because a hydride ion is transferred from the molecule being oxidized to the one being reduced. In the next few sections, we will learn about two important classes of coenzyme molecules that serve hydride ion acceptors (oxidizing agents) and hydride ion donors (reducing agents) in biochemical redox reactions.
Be careful not to confuse the terms hydrogenation and dehydrogenation with hydration and dehydration – the latter terms refer to the gain or loss of water, while the former terms refer to the gain or loss of hydrogen.
Many mechanistic patterns that we have already learned about in previous chapters will come into play again in this discussion, with the only variation being that here, a hydride ion will act as a nucleophile (in the hydrogenation direction) or as a leaving group (in the dehydrogenation direction). The key to understanding these reactions will be to understand how a hydride can act as a nucleophile or leaving group.
Nicotinamide adenine dinucleotide – a hydride transfer coenzyme
Although we are talking here about hydrides acting as nucleophiles and leaving groups, you already know that literal hydride ions are far too unstable to exist as discreet intermediates in the organic reactions of living cells (the pKapKa of H2H2, the conjugate acid of hydride, is about 35: a very weak acid, meaning hydride is a very strong base and not a reasonable species to propose for a biochemical reaction). As was alluded to earlier, biochemical hydrogenation/dehydrogenation steps require the participation of a specialized hydride transfer coenzyme. The most important of these is a molecule called nicotinamide adenine dinucleotide. The full structure of the oxidized form of this coenzyme, abbreviated NAD+NAD+, is shown below, with the active nicotinamide group colored blue. Because the redox chemistry occurs specifically at the nicotinamide ring (in blue in the figure below), typically the rest of the molecule is simply designated as an ‘R’ group.
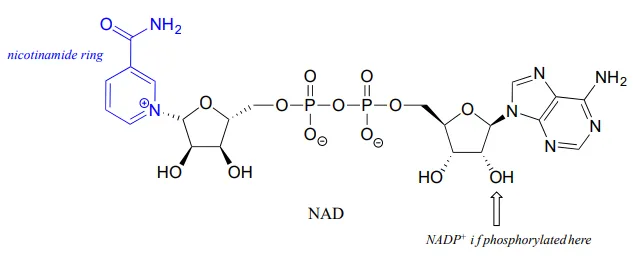
If the hydroxyl group indicated by the arrow is phosphorylated, the coenzyme is called NADP+NADP+. The phosphate is located far from the nicotinamide ring and does not participate directly in the hydride transfer function of the cofactor. It is, however, important in a larger metabolic context: as a general rule, redox enzymes involved in catabolism (the breakdown of large molecules) typically use the non-phosphorylated coenzyme, while those involved in anabolism (biosynthesis of large molecules from small precursors) use the phosphorylated coenzyme.
NAD+NAD+ and NADP+NADP+ both function in biochemical redox reactions as hydride acceptors: that is, as oxidizing agents. The reduced forms of the coenzyme, abbreviated NADHNADH and NADPHNADPH, serve as hydride donors: that is, as reducing agents.
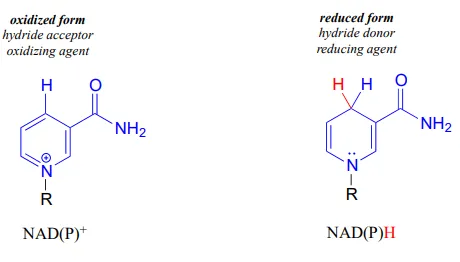
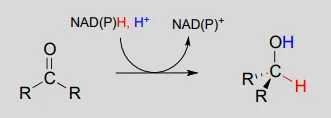
To understand how the nicotinamide coenzymes function in hydride transfer, let’s look at a general picture of a reversible, redox conversion from a ketone to a secondary alcohol. Mechanistically, the reaction we are about to see can be described as a nucleophilic addition to a carbonyl – a mechanism type we studied in chapter 10 – with the twist that the nucleophilic species is a hydride ion. At the beginning of the reaction cycle, both the ketone substrate and the NADHNADH cofactor are bound in the enzyme’s active site, and carbon #4 of the nicotinamide ring is positioned very close to the carbonyl carbon of the ketone.
NAD(P)HNAD(P)H-dependent hydrogenation (reduction) of a ketone
Mechanism:
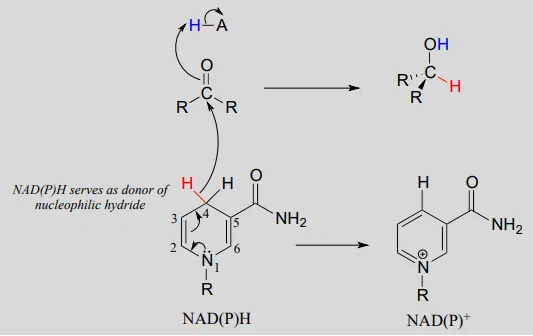
As an enzymatic group transfers a proton to the ketone oxygen, the carbonyl carbon loses electron density and becomes more electrophilic, and is attacked by a hydride from NADHNADH. Because carbon #4 of NADHNADH is bound in such close proximity to the electrophile, this step can occur without generating a free hydride ion intermediate – the two hydride electrons can be pictured as shifting from one carbon to another. Note the products of this reaction: the ketone (which accepted a hydride and a proton) has been reduced to an alcohol, and the NADHNADH cofactor (which donated a hydride) has been oxidized to NAD+NAD+.
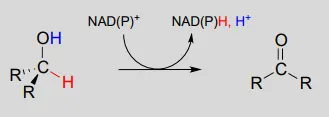
The dehydrogenation of an alcohol by NAD+NAD+ is simply the reverse of a ketone hydrogenation:
NAD(P)+NAD(P)+-dependent dehydrogenation (oxidation) of an alcohol
Mechanism:
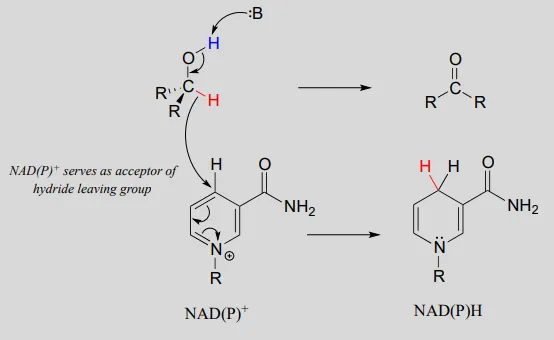
An enzymatic base positioned above the carbonyl removes a proton, and the electrons in the O−HO−H bond shift down and push out the hydride, which shifts over to carbon #4 of NAD+NAD+. Note that the same process with a primary alcohol would yield an aldehyde instead of a ketone.
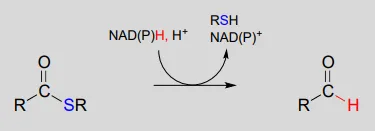
The nicotinamide coenzymes also serve as hydride donors/acceptors in the redox reactions interconverting carboxylic acid derivatives and aldehydes. Notice that these reactions can be thought of as nucleophilic acyl substitution reactions (chapter 11) in which the nucleophile or leaving group is a hydride ion.
NAD(P)H-dependent hydrogenation (reduction) of a thioester to an aldehyde:
Mechanism:
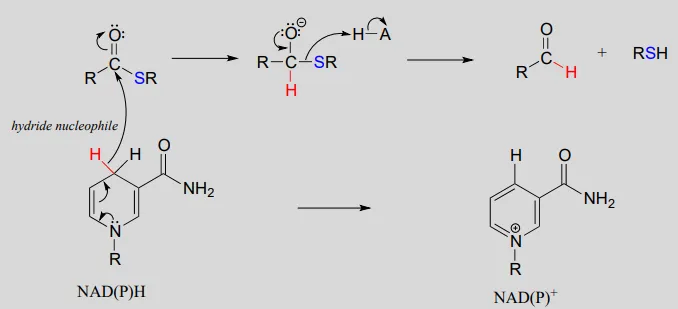
NAD(P)+NAD(P)+-dependent dehydrogenation (oxidation) of an aldehyde to a thioester:
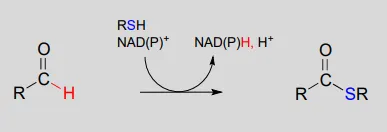
Mechanism:
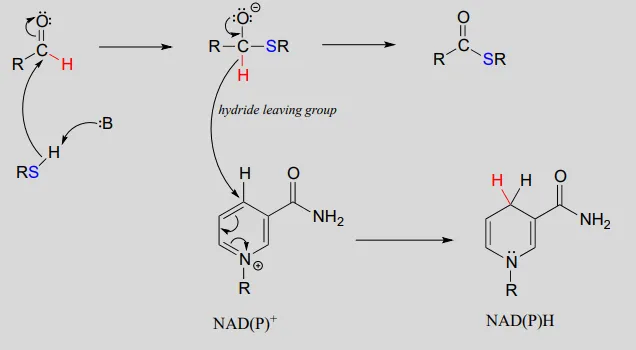
To simplify figures, hydrogenation and dehydrogenation reactions are often drawn with the role of the coenzyme abbreviated:
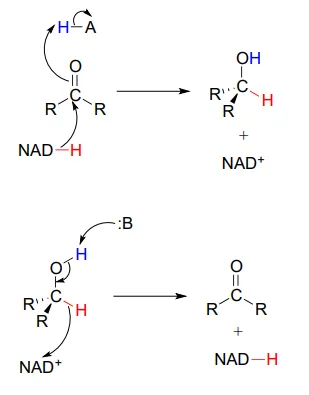
However, it is very important to make sure that you can remember and draw out the full mechanism, including the role of the coenzyme, in these types of reactions.
CAUTION
A very common error made by students learning how to draw biochemical redox mechanisms is to incorrectly show nicotinamide coenzymes acting as acids or bases. Remember: \(NADH\) and \(NADPH\) are hydride donors, NOT proton donors. \(NAD^+\) and \(NADP^+\) are hydride acceptors, NOT proton acceptors.
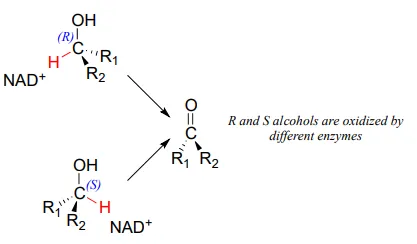
Stereochemistry of ketone hydrogenation
It should not surprise you that the stereochemical outcomes of enzymatic hydrogenation / dehydrogenation steps are very specific. Consider the hydrogenation of an asymmetric ketone: In the hydrogenation direction, attack by the hydride can occur from either the re or the si face of an asymmetric ketone (see section 10.1), leading specifically to the S or R alcohol.
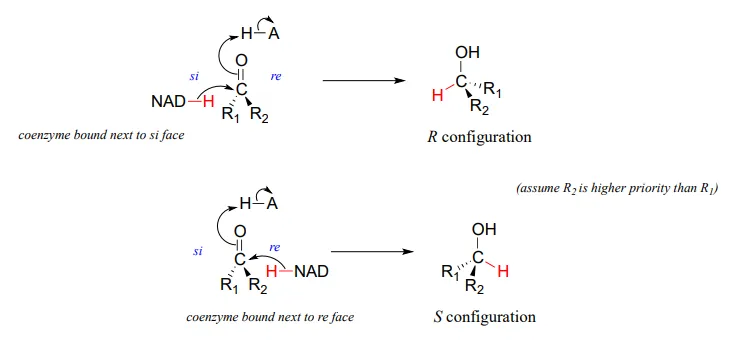
The stereochemical configuration of the product depends on which side of the ketone substrate the NAD(P)HNAD(P)H coenzyme is bound in the active site. Any given enzyme will catalyze its reaction with one of these two stereochemical outcomes, not both.
Stereochemical considerations apply in the dehydrogenase direction as well: in general, enzymes specifically catalyze the oxidation of either an R or S alcohol, but not both.
a. Draw the structure of the starting ketone in this reaction.
b. Which face of the ketone is the coenzyme positioned next to in the active site of the enzyme?
Examples of biochemical carbonyl/imine hydrogenation
Now that we have covered the basics, let’s look at some real examples of hydrogenation and dehydrogenation reactions.
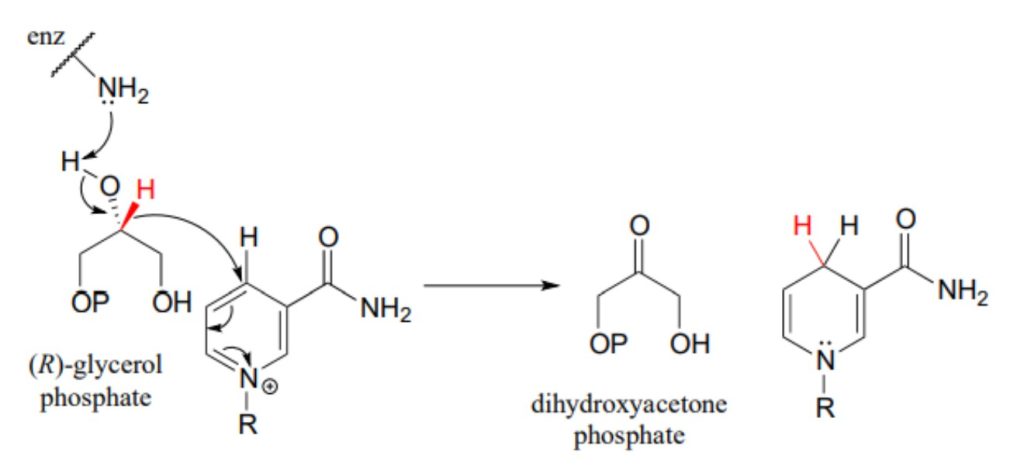
Glycerol phosphate dehydrogenase (EC 1.1.1.8) catalyzes one of the final chemical steps in the breakdown of fat molecules. The enzyme specifically oxidizes (R)-glycerol phosphate to dihydroxyacetone phosphate. (S)-glycerol phosphate is not a substrate for this enzyme. J. Mol. Biol. 2006, 357, 858 (human crystal structure)
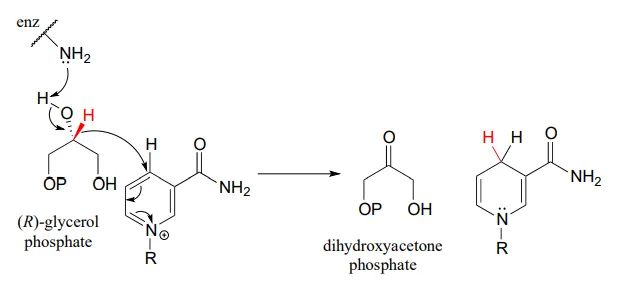
The reverse reaction (catalyzed by the same enzyme) converts dihydroxyacetone phosphate to (R)-glycerol phosphate, which serves as a starting point for the biosynthesis of membrane lipid molecules (see section 1.3).
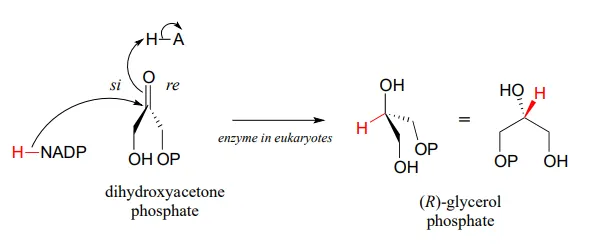
While the cell membranes of animals, plants, and bacteria are made from lipids with the R stereochemistry exclusively, archaeal microbes (the so-called ‘third kingdom of life) are distinguished in part by the S stereochemistry of their membranes.
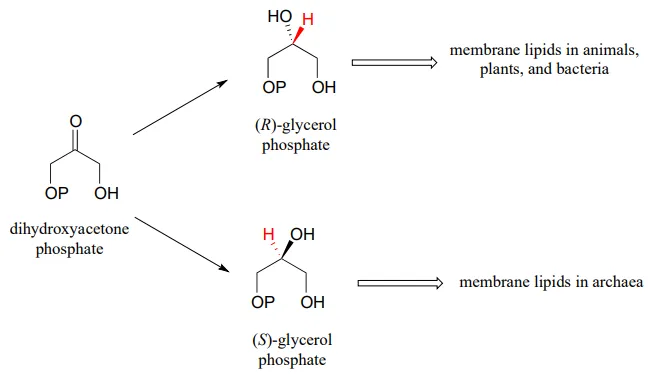
fig 27
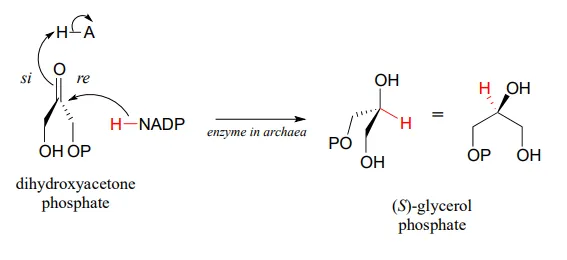
Archaea have an enzyme that catalyzes hydrogenation of dihydroxyacetone with the opposite stereochemistry compared to the analogous enzyme in bacteria and eukaryotes. This archaeal enzyme was identified and isolated in 1997.
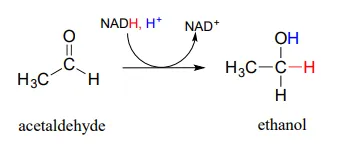
In a reaction that is relevant to people who enjoy the occasional ‘adult beverage’, an NADHNADH-dependent enzyme (EC 1.1.1.1) in brewer’s yeast produces ethanol by reducing acetaldehyde. This is the final step in the process by which yeast ferment glucose to ethanol.
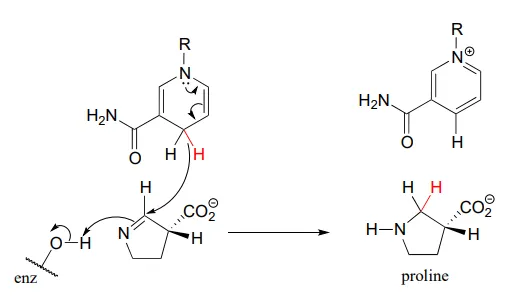
The reaction below, which is the final step in the biosynthesis of proline (EC 1.5.1.2), is an example of an enzymatic reduction of an imine to an amine. J. Mol. Biol. 2005, 354, 91.
This step in the breakdown of the amino acids glutamate (EC 1.4.1.2) provides an example of the oxidation of an amine to an imine: Structure 1999, 7, 769.
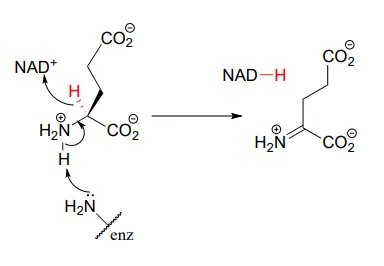
The ‘double reduction’ reaction below (EC 1.1.1.34) is part of the isoprenoid biosynthetic pathway, which eventually leads to cholesterol in humans.
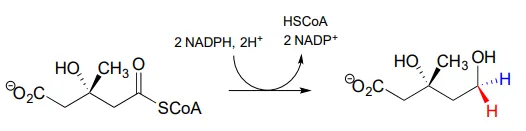
In this reaction, a thioester is first reduced to an aldehyde in steps 1a and 1b:
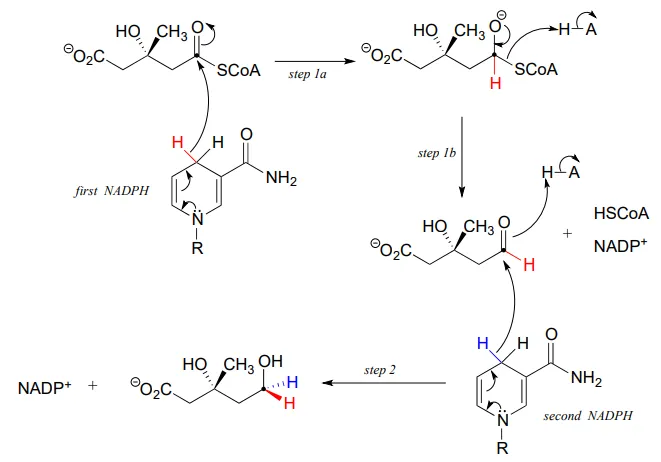
Then in step 2, the aldehyde is in turn reduced by the same enzyme (and a second NADPHNADPH that enters the active site) to a primary alcohol. This enzyme is inhibited by atorvastatin and other members of the statin family of cholesterol-lowering drugs. Atorvastatin, marketed under the trade name Lipitor by Pfizer, is one of the all-time best-selling prescription medications.
Recall from chapter 11 that carboxylates are not reactive in acyl substitution steps, so it follows that they cannot be directly reduced to aldehydes by an enzyme in the same way that thioesters can. However, a carboxylate can be converted to its ‘activated’ acyl phosphate form (section 11.4), which can then be hydrogenated. An example of this is found in a two-reaction sequence found in amino acid metabolism (EC 2.7.2.11; EC1.2.1.41).
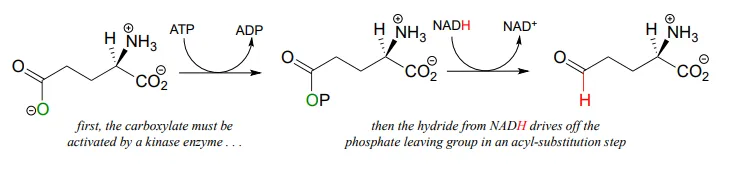
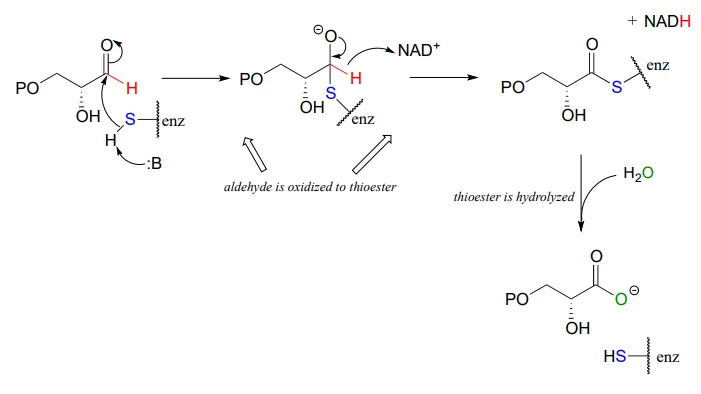
Glyceraldehyde-3-phosphate dehydrogenase (EC 1.2.1.12) , a key enzyme in the glycolysis pathway, provides an example of the oxidation of an aldehyde to a thioester, in this case a thioester linkage between the substrate and a cysteine residue in the enzyme’s active site. In the second phase of the reaction, the thioester intermediate is hydrolyzed to free the carboxylate product.
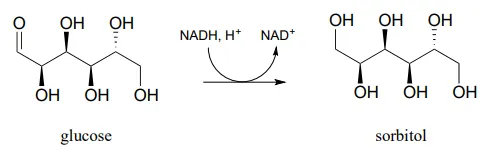
Draw a likely mechanism for the conversion of glucose to sorbitol, a process that occurs in the liver. Do not abbreviate the nicotinamide ring structure.
Reduction of ketones and aldehydes in the laboratory
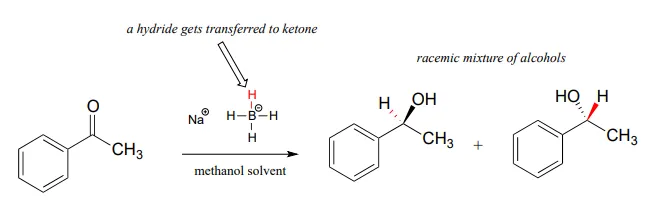
Although our focus in this book is biological organic reactions, it is interesting to note that synthetic organic chemists frequently perform hydrogenation reactions in the lab that are similar in many respects to the NAD(P)H-dependent reactions that we have just finished studying. A reagent called sodium borohydride (NaBH4NaBH4) is very commonly used, often in methanol solvent, to reduce ketones and aldehydes to alcohols. The reagent is essentially a laboratory equivalent of NADHNADH (or NADPHNADPH): it serves as a source of nucleophilic hydride ions. Sodium borohydride is a selective reagent in the sense that it will reduce ketones and aldehydes but not carboxylic acid derivatives such as esters (recall from section 11.2 and section 11.3 that the carbonyl carbons of carboxylic acid derivatives are less potent electrophiles than the carbonyl carbons of ketones and aldehydes). Unlike the enzymatic hydrogenation reactions we saw earlier, the reduction of asymmetric ketones with sodium borohydride usually results in a 50:50 racemic mixture of the R and S enantiomers of the alcohol product.
Synthetic organic chemists have at their disposal a wide range of other reducing and oxidizing reagents with varying specificities and properties, many of which you will learn about if you take a course in laboratory synthesis.